Where Dreams Find Direction


Growth. Direction. Excellence.
South College is a private college offering more than 100 Programs and Concentrations. We have locations in Tennessee, North Carolina, Georgia, Indiana, Pennsylvania, and Florida, plus Online and Competency Based Education (CBE) as well. Specialized focus is placed on Associate, Bachelor’s, Master’s, Educational Specialist, and Doctoral degree programs in Healthcare, Business, Technology, Education, and Legal Studies, as well as select Certificate programs. Modern labs and classrooms with relevant technology and equipment power exceptional hands-on and real-world focus for the serious student.
To serve students and assist in their exploration and discovery during their educational pursuits, we are proud to participate in many scholarship and grant opportunities. For our Tennessee students, the Tennessee Promise Scholarship, Tennessee Reconnect Scholarship, and Hope Scholarship are available to those who qualify. In Indiana, the 21st Century Scholars Program and the Frank O’Bannon Grant are available to students who qualify. For our Florida students, the Florida EASE Grant and the Florida Bright Futures Scholarship are available to those who qualify. Our Georgia, North Carolina, and Pennsylvania students have the opportunity to qualify for South College Promise. The Yellow Ribbon Scholarship is also currently offered for all VA Education benefit approved locations.
Explore 100+ Programs & Concentrations in a location near you
100+ Programs & Concentrations
South College offers more than 100 Programs and Concentrations at the Certificate, Associate, Bachelor’s, Master’s, Educational Specialist, and Doctoral levels at our campuses in Knoxville (two locations), Nashville, Asheville, Atlanta, Indianapolis, Pittsburgh, and Orlando, as well as both Online and Competency Based Education (CBE). We are constantly developing new academic programs based on local, regional, and national trends. Each curriculum is designed is to promote development of knowledge and skills in the associated discipline as we challenge and support our students. We’re here with you every step of the way, starting with the admissions process!
Asheville
Atlanta
Indianapolis
Knoxville
Nashville
Orlando
Pittsburgh
Online
CBE – Competency Based Education
Asheville
Set in the scenic mountains of western North Carolina, the South College Asheville campus has helped many students and professionals pursue success with relevant academic programs. Our facility combines a classic setting with modern detailing and ample parking. It houses all of our campus services, plus your classes, in one convenient location—one of the most beautiful campuses found among Asheville colleges. Start working toward your career goals today! We’re ready to help with programs designed to help you pursue your goals, most of which have no wait list!
AshevilleAtlanta
At the South College Atlanta Campus, students are provided with the opportunity to pursue educational programs that brings talents and skills together for a meaningful future impact. This modern facility, conveniently located on the I-85 corridor, inside the Perimeter, houses all your needs for educational growth in one convenient location. You’re ready to start working towards your goals today – and we’re ready to help. Once your enrollment is complete, you’ll be able to join the next class of South College students and start turning your dreams into your future!
AtlantaIndianapolis
South College’s beautiful, modern Indianapolis Campus provides opportunities for students to pursue educational programs designed to help prepare for success in their future goals. Our practical education will help you to develop your talent, skills, and knowledge that promote a meaningful impact on your future. Throughout your time as a student, our Indianapolis faculty and staff will be available to give you one-on-one support and direction to help you pursue your goals. You’re ready to start working towards your goals today – and we’re ready to help. Once you enroll, you’ll be able to join the next class of South College students and start turning your dreams into reality!
IndianapolisKnoxville
South College has 2 convenient locations in Knoxville – the Main Campus located on Lonas Drive and the Parkside Campus located on Goody’s Lane. The Lonas Campus includes modern lecture classrooms, specialty laboratories equipped with relevant equipment, a Barnes & Noble bookstore, and the library/resource center. Ample parking is available for students, faculty, and staff. Knoxville-Lonas is also the home of our Corporate offices. You’re ready to start working towards your professional goals today – and we’re ready to help. That’s why many of our programs have no waitlist. Once your enrollment is complete, you’ll be able to join the next class of South College students, and start working toward turning your dreams into your reality!
KnoxvilleNashville
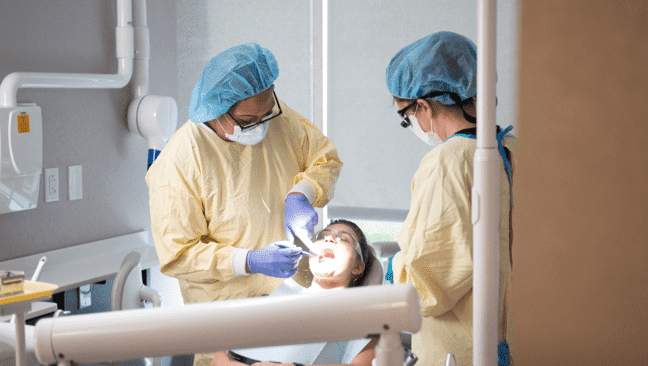
Across from the airport on I-40, South College’s Nashville Campus includes modern classrooms, laboratories, and other conveniences for students and faculty including a Barnes & Noble Bookstore, a public-facing Dental Clinic, large Student Centers, and more. The campus has parking available and houses all of your classrooms, study rooms, labs, and resource center in one convenient location. The building features high-tech SIM-labs and other healthcare technology including computers, projectors, x-ray machines, and much more. You’re ready to start working towards your professional goals today – and we’re ready to help. That’s why many of our programs have no wait-list. Once your enrollment is complete, you’ll be able to join the next class of South College students, and start turning your dreams into your reality!
NashvilleOrlando
At South College’s Orlando Campus, our associate degree, bachelor’s degree, and certificate programs are designed to help prepare students to pursue their professional goals. Our practical, career-focused approach to education is designed to teach you relevant skills and equip you to make a meaningful impact on your community and the professional field of your choosing. To help you succeed, our experienced faculty and staff provide one-on-one attention and mentorship and our facilities feature modern equipment and labs. Get started working towards your professional goals today at the South College Orlando campus! Once you enroll, you’ll discover a friendly, supportive environment with the resources you need to work toward turning your dreams into a reality.
OrlandoPittsburgh
At South College’s brand new Pittsburgh campus, our certificate and degree programs are designed to help prepare you to pursue your goals. With a practical, focused approach to education, we work to teach you to think critically, communicate effectively, and apply your knowledge to real world situations. We are committed to helping you develop foundational knowledge and skills with the latest equipment and technology. To help you succeed, our friendly and experienced instructors and staff offer personal attention and mentorship to every student. We also understand the importance of convenience and flexibility, with opportunities for students to take select courses online or on campus. Take the next step today by requesting information about the South College Pittsburgh campus.
PittsburghOnline
South College Online is where you take your first steps toward pursuing your personal goals and working toward opportunities to turn your dreams into a satisfying direction. It’s also where you can choose between 90+ online degree programs, concentrations and certificate programs. Our online programs provide pathways to earn academic credentials at a variety of levels including certificate, associate, bachelor’s, master’s, educational specialist, or even a doctoral degree. In these programs, you will enjoy the high-quality, high-impact education we have provided for over 141 years – but on your own schedule, and from anywhere you like – through online courses.
OnlineCBE – Competency Based Education
The South College Competency-Based delivery modality (or CBE) is an innovative educational approach that creates opportunity for students to evidence mastery of required content at their own pace in their own surroundings . This delivery method creates a unique environment for self-starters, working professionals, and students who prefer to learn at their own speed.
CBE – Competency Based EducationYour Journey at South College
When you start your new path at South College, you’ll join students from all walks of life and at all stages in their lives. Many students are going back to school to finish degrees, while others are pursuing elevated education that will help improve their potential in their current roles or chasing a new direction.
Some students are recent high school graduates starting college for the first time, and still many others are veterans seeking their next opportunity after military service. However, all of our students are here for the experienced faculty and staff members, modern, high-tech labs, and market-based programs that make South College a thriving educational community and challenging learning environment for the serious student. We are committed to providing intellectual, social, and professional development to a diverse student body.
Looking for More Options?
Don’t live near one of our 8 campuses? We’ve got you covered!
Experience online learning on your own terms; your timeframe and flexibility with online classes!
We have two unique pathways to experience education flexibility; Online and Competency Based Education (CBE) programs.
South College Online is where you take your first steps toward pursuing your personal goals and working toward opportunities to turn your dreams into a satisfying direction. It’s also where you can choose between 100+ online degree programs, concentrations, and certificate programs.
With the self-pace of the Competency Based Education model and the flexibility to study anywhere you are, you will be able to explore a variety of Master’s, Doctoral, and Post-Doctoral Certificate programs in business, education, information technology, ministry, and more! You will also receive the benefit of a supportive community of a faculty mentor, advisors, and staff who will partner with you in your pursuit of academic excellence. This delivery method creates a unique environment for self-starters, working professionals, and students who prefer to learn at their own speed.
Affordability & Tuition Assistance
Financial Aid is available to those who qualify. Meet personally with a financial aid advisor and discover the financial resources through government aid, grants, and scholarships that may be available for you.


Accreditation
Frequently Asked Questions
South College participates in a range of financial aid programs. Financial Aid is available to students who qualify and the financial aid staff work individually with students to determine eligibility as they seek the economic assistance they need to pay for college. Aid is available in the form of scholarships, grants and awards, work-study programs, and loans. We’re happy to participate in numerous federal, state, and private student aid programs, and to offer funding directly. South College is a proud participant in the Tennessee Promise Scholarship, Hope Scholarship, Florida Bright Futures Scholarship, 21st Century Scholarship, South College Promise, and Yellow Ribbon Scholarship programs.
Go to Financial AidSouth College provides quality, accelerated educational programs in a variety of career fields. We hope that through the excellence of our programs you will be able to become a more qualified and marketable professional. Our tuition and fees are based on the operational costs of our institution as we strive to excel and advance education to new innovative and instructional heights. As there are various tuition schedules for our degrees, certificates and concentrations we encourage you to see which tuition schedule fits your desired program. Please see Tuition & Fees and then click either Campus Tuition or Online Tuition.
Tuition & FeesYes, South College is a private institution providing a longstanding tradition of leadership in education with thousands of graduates in nursing, business, criminal justice, health science, pharmacy, and legal studies.
Yes, South College is accredited by several governing bodies as a private institution providing a tradition of leadership in education with thousands of graduates in nursing, business, criminal justice, health science, pharmacy, and legal studies, and more. Click below to see all Accreditation information.
AccreditationsUnfortunately, South College does not offer housing and there are no dormitory facilities at any South College campus. Students who are not within commuting distance of one of our campuses must secure their own residence. Resources are available from the Department of Student Services.
There are several avenues for general admission as an Undergraduate. Please click the link below to explore these requirements. Those applying to graduate programs should review admission and application requirements in the catalog section for each program.
Admission RequirementsYes. Through this program, qualified high school students may pursue collegiate coursework either online or in person while completing their high school requirements. Students accepted for dual enrollment can enroll part-time or full-time at any of our locations provided they meet course prerequisites and receive permission from their high school principal or counselor. Qualified students residing in Tennessee are eligible for the Tennessee Dual Enrollment Grant which fully funds their dual enrollment courses at South College. The South College Promise is a tuition assistance grant funded directly by South College and awarded to qualified high school students who wish to pursue dual enrollment courses at our Asheville, Atlanta, Orlando, Pittsburgh and Indianapolis campuses.
Yes, South College participates in scholarships available to those who qualify including the Tennessee Education Lottery Scholarship, Tennessee Promise, HOPE Scholarship, Tennessee Reconnect, Florida Bright Futures Scholarship, Indiana 21st Century Scholarship, and the Yellow Ribbon Promise to name a few. South College also offers several different Grant programs such as those for active military and veterans and those in teacher education programs.
Financial Aid OptionsSouth College is pleased to provide Microsoft Office Pro Plus (Word, Excel, PowerPoint, Access, Publisher, OneNote, and InfoPath) to all students enrolled in credit courses at no cost via the Microsoft Student Advantage program! South College, and most of your Instructors, use Microsoft Office programs (Word, Excel, PowerPoint, etc.). To make sure your Instructors can open and grade your assignments, it’s easier to use Microsoft Office programs to create and submit your assignments. You also will receive free downloads of needed applications such as Adobe Acrobat Reader, Quick Time, Windows Media Player and more. Help Desk Assistance is available too.
The South College Military Grant is available to all active-duty military members, veterans with honorable or general discharge status, and military spouses who are not receiving 100% coverage of tuition and fees under any VA educational benefit and/or state, federal, institutional, or private grant or scholarship program. South College has also been a proud participant in the Yellow Ribbon program since 2011. We have other state-specific military grants and programs available to those that qualify. Please ask our Financial Aid team what options you may have within your state of residence.
Military and Veterans Services